بیماری فنیل کتونوری چیست؟
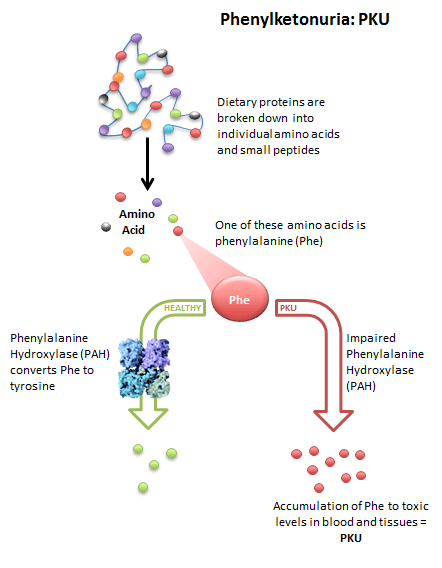
یماری فنیل کتونوری یک نقص متابولیکی مادرزادی نادر است. اختلال اصلی در این بیماری، تجمع اسید آمینه فنیل آلانین در مایعات بدن و سیستم عصبی است. تجمع این اسید آمینه به دلیل عدم وجود آنزیم مورد نیاز برای تبدیل فنیل آلانین به تیروزین رخ میدهد. تجمع غیرطبیعی این اسید آمینه در بدن کودک، خطرناک است و منجر به بروز اختلالاتی در مغز و پوست میشود. تجمع اسید آمینه فنیل آلانین در بدن کودک باعث ناتوانی ذهنی (عقب ماندگی ذهنی) میشود.
بیماریزایی:
فنیل آلانین پس از ورود به بدن توسط آنزیمی به نام فنیل آلانین هیدروکسیلاز شکسته و به تیروزین تبدیل میشود. کوآنزیم این واکنش، تتراهیدروبیوپترن (BH4) میباشد. سپس تیروزین شکسته شده و به مواد متعددی از جمله رنگدانه پوست و مو تبدیل شده و متابولیتهای نهایی آن از بدن دفع میشود. پس چنانچه آنزیم «فنیل آلانین هیدروکسیلاز» که فقط در کبد ساخته میشود به دلیل اختلالات ژنی وجود نداشته باشد «فنیل آلانین» وارد شده به بدن، در بافتهای مختلف از جمله مغز تجمع یافته و سبب آسیبهای متعددی به بافت مغز میشود.
شیوه انتقال:
ن بیماری مانند تالاسمی به صورت اتوزوم مغلوب به ارث میرسد؛ و این یعنی جنسیت فرد در ابتلا به آن نقش مستقیم و ویژهای ندارد، ژن این بیماری بر روی کروموزوم ۱۲ قرار گرفتهاست. چنانچه والدین هردو حامل این ژن باشند و خود سالم باشند (که معمولاً در ازدواجهای خویشاوندی این احتمال بالاتر است) در تولد هر فرزندانشان ۲۵ درصد احتمال ابتلا به فنیل کتونوریا وجود دارد. البته از آن جا آزمایش تشخیص این بیماری بسیار آسان و کم هزینه است، در بیمارستانها و زایشگاههای بسیاری از کشورهای پیشرفته، همه نوزادان را از نظر دارا بودن عامل این بیماری آزمون میکنند. بهطور کلی توصیه میشود افراد پیش از هرگونه اقدام در زمینه ازدواج به مشاوران ژنتیک حتماً مراجعه بکنند به ویژه افرادی که در خانواده آنان سابقه ابتلا به هر یک از بیماریهای ژنتیکی وجود دارد.
این بیماری در دختران و پسران به یک نسبت مشاهده می شود. هر فرد دارای دو ژن است که دستور ساخت آنزیم PAH را میدهد. در کودکان PKU هیچ یک از این دو ژن به درستی عمل نمی کنند. این کودکان هر یک از ژنهای معیوب را از یکی از والدین خود به ارث برده اند. به این نوع توارث، وراثت اتوزومال مغلوب می گویند. در والدین این کودکان، به ندرت اختلالی مشاهده می شود. در عوض هر یک از والدین یک تک ژن معیوب برای PKU دارد. این والدین ناقل نامیده می شوند. ناقلین علامت بیماری را بروز نمی دهند زیرا دیگر ژن آنها سالم است. وقتی هر دو والدین ناقل باشند، ۲۵% احتمال تولد کودک کاملا سالم ،۵۰% احتمال تولد کودک ناقل همچون والدین و ۲۵% احتمال تولد کودک مبتلا به PKU در هر بارداری وجود دارد. مشاوران ژنتیک برای پاسخگویی به سوالات خانواده ها در دسترس می باشند. این مشاوران نحوه توارث بیماری را توضیح خواهند داد و همچنین راهنمای خانواده ها برای بارداری بعدی و همچنین مشاور آنان برای انجام تستهای تشخیصی برای دیگر اعضای خانواده خواهند بود.
تنها راه سنتز تیروزین در انسان از همین طریق است. در افراد مبتلا به فنیل کتونوری تیروزین، اسید آمینه ضروری میشود و باید از غذا تأمین شود. در این افراد فنیل آلانین خون نیز به میزان زیادی بالا میرود. در افراد طبیعی مقدار کمی از فنیل آلانین، به فنیل پیروات، فنیل استات و فنیل لاکتات تبدیل میشود اما در افراد مبتلا به فنیل کتونوری به علت بالا بودن میزان فنیل آلانین، مقدار زیادی از این فنیل کتونها تولید شده که وارد خون و ادرار میشوند. مقدار زیاد فنیل پیروات در خون، از عمل آنزیم پیروات دکربوکسیلاز در مغز به صورت آلوستریک، جلوگیری میکند. این عمل به نوبه خود در تشکیل میلین اشکال ایجاد میکند و منجر به عقب ماندگی ذهنی میشود. یکی دیگر از دلایل عقب ماندگی ذهنی در افراد مبتلا به فنیل کتونوری کاهش تولید نوروترانسمیترهایی مانند دوپامین میباشد. تیروزین پیش ساز سنتز این نوروترانسمیترها است. در این بیماران مقدار دوپامین و سروتونین در ادرار کمتر از حد طبیعی است. مصرف غذاهای پروتئینی از جمله شیرخشکهای معمولی و به میزان کمتر شیرمادر باعث افزایش شدید غلظت خونی فنیل آلانین و تجمع آن در بدن، اختلال در تکامل مغز و اعصاب و در نهایت ضایعه مغزی و عقب ماندگی ذهنی پایدار در مبتلایان میشود. تجمع فنیل آلانین و مشتقات غیرطبیعی آن در نسوج مختلف، فعالیت سلولها، به خصوص سلولهای سیستم عصبی را تحت تأثیر قرار داده و از رشد آن جلوگیری میکند.
این ضایعات متأسفانه در هفتههای اول تولد، نشانههای واضحی ندارند در نتیجه بیماری بموقع تشخیص داده نشده و درمان آن به تأخیر میافتد. در چنین دوران بحرانی که حیاتیترین مرحله شکلگیری و تکامل مغز کودک است، تغذیه با شیرمادر یا شیرخشک معمولی یا هر ماده پروتئینی دیگر اگر ادامه یابد منجر به ضایعه عصبی میشود و توان هوشی و قدرت ادراک کودک به تدریج تضعیف میشود و در نهایت منجر به معلولیت شدید ذهنی میگردد. متأسفانه گذشت زمان، شانس طلایی درمان را از بین برده و رژیم غذایی و درمانهای بعدی، دیگر نتیجه مطلوب را نخواهند داشت.
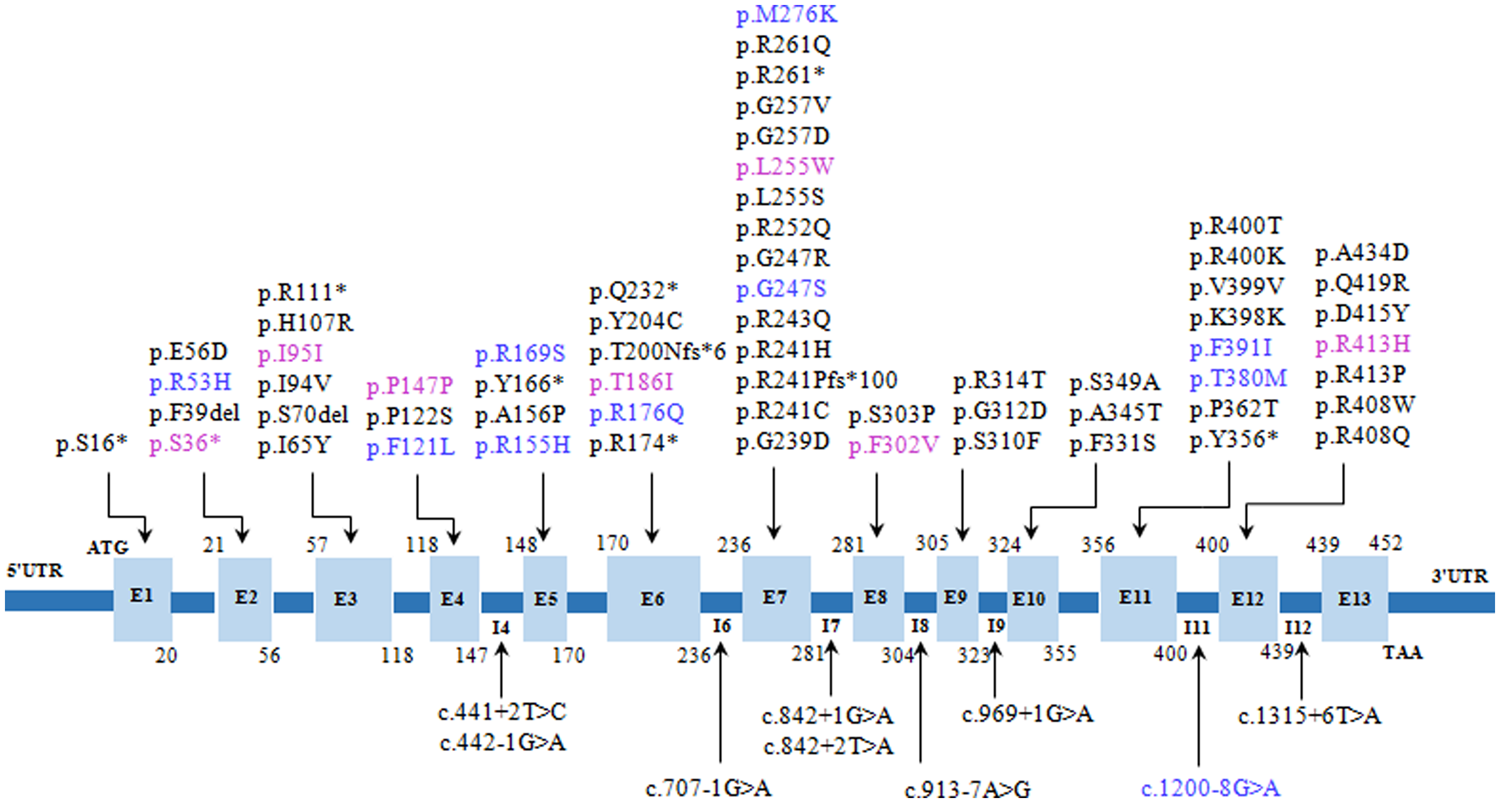
جهش های شناسایی شده در ژن PAH